

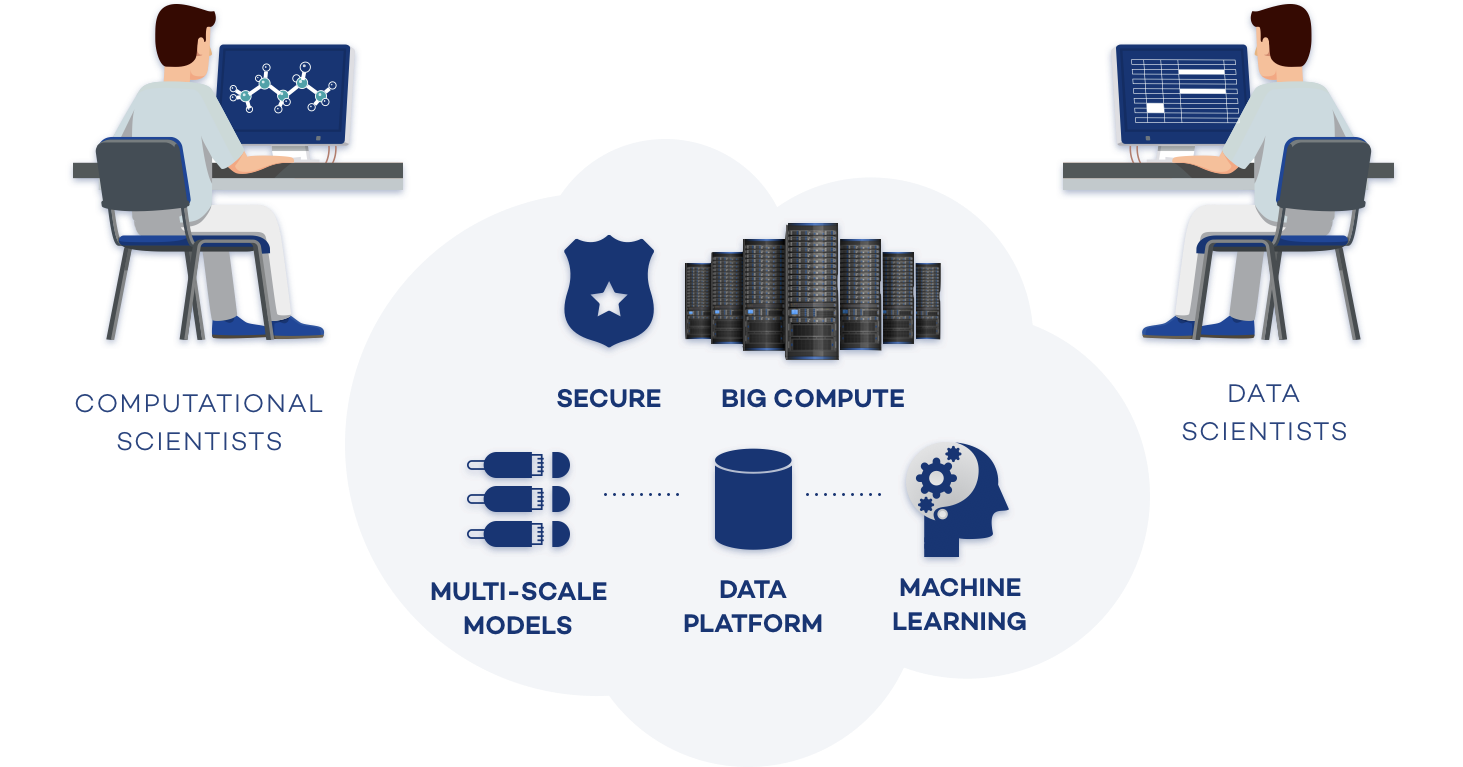
Exabyte.io is a cloud-native digital materials R&D platform. Design structures, run simulations, and build models online alongside scientists on your team and worldwide.
READY FOR MATERIAL GAINS?
NEW
materials
microprocessors
data storage
solar cells
batteries
catalysts
alloys
polymers
chemicals
Digitally transform R&D practices for new materials and chemicals
with our machine intelligence.
THE EXABYTE.IO PLATFORM
Exabyte.io helps chemists and materials scientists organize their work and collaborate in a single easy-to-use cloud environment, allowing to rapidly learn and deploy a variety of modeling tools able to accelerate the R&D phase and provide the foundation for faster product development.

0
Simulation Workflows0
Materials Stored0
Calculations Run0
Properties PredictedPlATFORM CAPABILITIES
From idea to result - let's get there faster together!
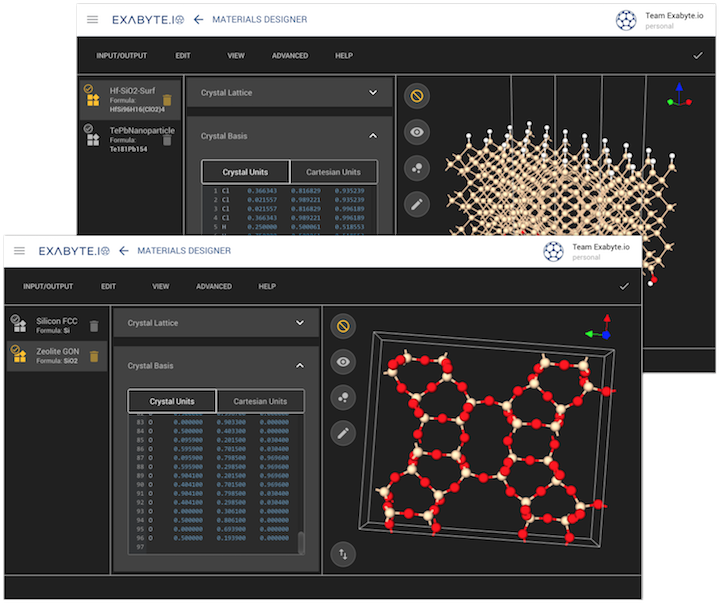
in-browser integrated environment for materials design
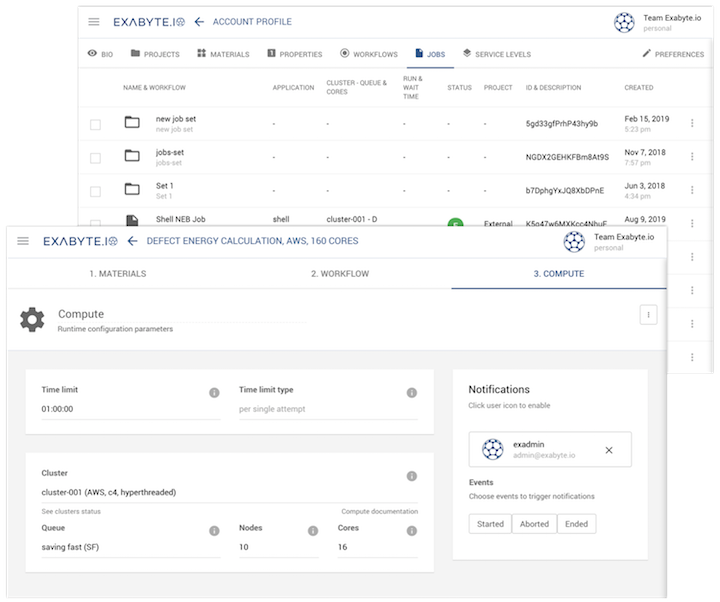
fast and secure cloud high-performance computing
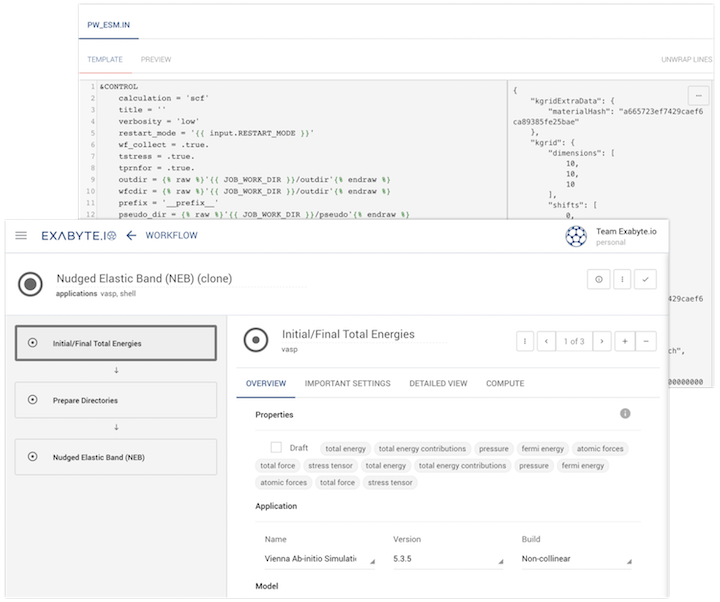
web-based designer for inter-operable modeling workflows
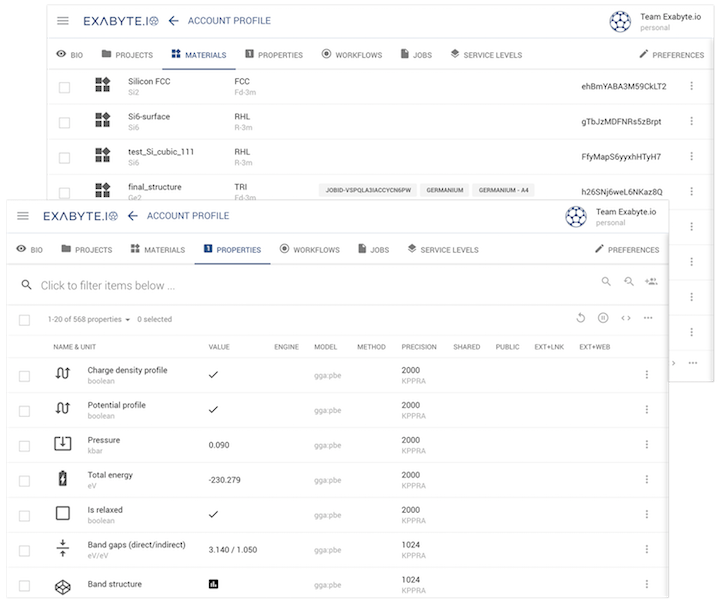
open-source data standards organizing materials information
advanced data analytics and machine learning infrastructure
secure collaboration within and between accounts
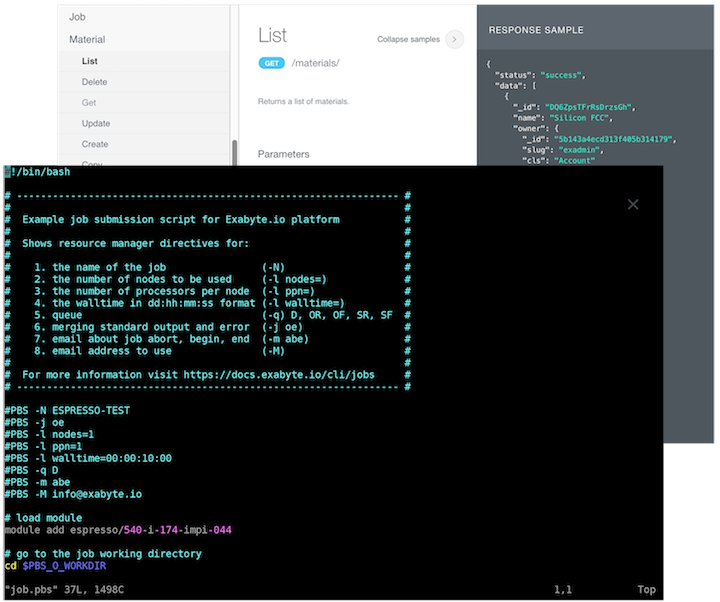
command-line interface, remote desktop, RESTful API access options for experts
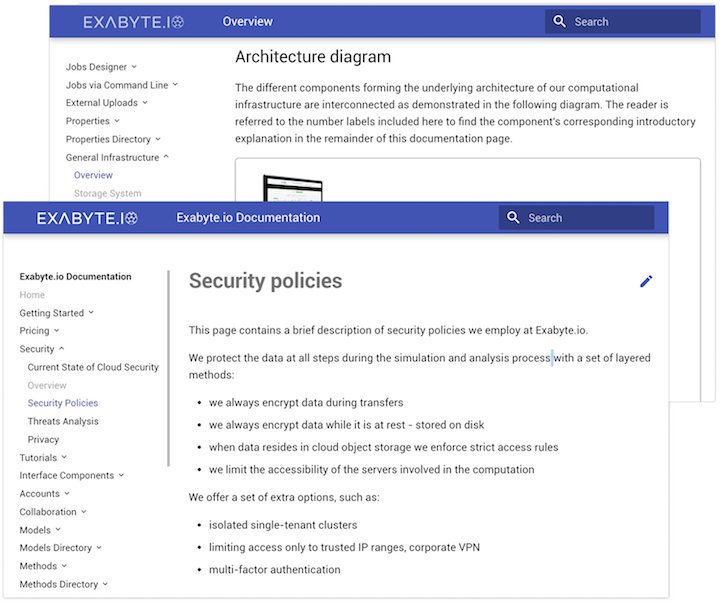
encrypted data at rest and in-transfer, network partitioning, and more for maximum security
related information:
CUSTOMER STORIES
"Exabyte.io allowed us to significantly accelerate our
research and development efforts by leveraging state-of-the-art advancements in
materials simulations and high performance computing in the cloud."
Dr. Sergey Barabash, Materials & Device Simulations,
Intermolecular, Inc.
"Exabyte.io lets us improve speed and
accuracy of the modeling techniques we deploy and helps organize the resulting
data. Accessible and collaborative interface allows us to train new users in
nanoscale simulations much faster."
Dr. Kazuki Mori, Science and Engineering Systems Division,
ITOCHU Techno-solutions, Inc.
"With Exabyte.io we have top-tier computational resources and support
in a very cost-efficient manner. This helps us
reduce the time required to educate new group members in
performing state-of-the-art research in molecular modeling."
Dr. Jun Koyanagi, Dept. of Materials Science and Technology,
Tokyo University of Science
FASTER MATERIALS R&D FOR
SEMICONDUCTOR
Emerging memory technology and TCAD: speed up material selection and process optimization for next-generation devices.
MANUFACTURING
Predict and assess the stability of lightweight metallic alloys, thin-film oxide compounds, composite materials.
ENERGY
IT engineers, materials- and data scientists collaborate to speed up the design of new compounds from atoms up.
Material progress - it's elemental
Exabyte.io modeling platform paves the way
Materials modeling today is an esoteric, albeit critical, discipline, hamstrung by a proliferation of specialized point tools, computational complexity, lack of standards, and an unmanaged explosion of data.
In order for the promise of materials modeling to be realized, the technology has to become FAIR: producing Findable data, Accessibe and easy-to-use, with Inter-operable tools, and having Re-usable well-defined workflows. We believe these goals are attainable now - and are proving it, with examples listed here.
Here is what we believe:
- Better modeling cuts the time-to-market for new materials and resulting products in many application areas
- Researchers must be able to rapidly learn and adopt any state-of-the-art modeling tools, whether they are intimately familiar with the tool or not
- Simulations deliver the most precise answers when modelers and experimentalists collaborate efficiently, using agreed standards
- Cloud computing is taking the lead in performance, scalability, and security for high-performance computing (HPC) workflows, including materials modeling
This is why Exabyte.io is building a cloud-native, modular, accessible and collaborative modeling platform.
Cloud-native
Cloud is the fastest-growing segment of HPC, and for good reasons. The vendors have the resources to offer instant access and scalability on the leading current technology. They also know security is central to their business and can afford the most reliable solutions, which is why companies now run critical functions such as HR, payroll, and finance in the cloud. The cloud service providers' lead will only extend over time helping to solve the computational complexity of materials modeling.
Modular and Accessible
Because of the computational complexity, no single software provider offers best-in-class tools across all scales and physical-model types. However researchers want the flexibility to use the optimum tool for a specific analysis, without having to learn yet another new language and data format. A plug-in modular architecture allows researchers to pick the best combination of tools, and get the job done faster. Data standards and an intuitive interface control the ever-growing data and cut learning time, making the discipline less esoteric.
Collaborative
The way to improve any R&D process is to first define it and allow great minds to participate, build on what has been learned already, then provide feedback from the results achieved so the process can be improved. This means scientists need to be able to review and comment on each other's work, to define and refine workflows, and to engage experimentalists to evaluate the results. Instead of having modelers and experimentalists work independently and continue speaking different languages, exchanging information regularly lets teams achieve R&D goals faster and more accurately.
Summary
Materials modeling has the potential to speed the discovery of extraordinary new materials for many industries and applications. To do so, it must be set free from the ivory tower and in-house supercomputers, and become more widely accessible, easier to use, faster, and more precise.
This is Materials Modeling 2.0.
Do you share our vision? We'd love to hear what you think.
The company
Founded in 2015, Exabyte.io is a cloud-based software company headquartered in San Francisco, California. It enables materials scientists and engineers in academic and industrial sectors, including energy, semiconductor, and manufacturing, to rapidly adopt and deploy a wide variety of modeling techniques to develop new materials faster.
How we are different.Exabyte.io is the only end-to-end materials modeling platform. It is architected to consolidate any state-of-the-art simulation tools and data, allowing its users to design new compounds and run physics-based and machine-learning models. The purpose-built platform lets customers increase the impact of high-end simulations and accelerate R&D.
Why it matters.Just as computer-aided design revolutionized manufacturing in the past, software-driven approaches are transforming the $40B materials R&D industry today, enabling product design from atoms up. Global 2000 companies and universities worldwide use Exabyte.io establishing it as the next standard in materials modeling.
Leadership Team

Timur Bazhirov
Founder/CEO
Timur established Exabyte.io in 2015 after obtaining a physics Ph.D. degree at UC Berkeley. There he worked with Marvin Cohen on first-principles studies of materials, publishing a handful of peer-reviewed articles and one day realizing that the way we do materials modeling needs to be modernized. Timur graduated with honors from Moscow Institute of Physics and Technology, speaks Russian and Tatar, and can often be seen cycling up mt. Tamalpais.

James Dean
Computational Scientist
James defended his PhD in chemical engineering at the University of Pittsburgh in 2020. His research in the lab of Giannis Mpourmpakis leveraged computational chemistry and machine learning to enable the high-throughput screening of nanoparticle catalysts, and delivered first-of-their-kind predictive models of nanoparticle physical properties. When not tackling a new problem, his hobbies include music, computer graphics, and flight.
Advisory Board

Marvin Cohen
Scientific Advisor
Professor of Physics at UC Berkeley. Author of over 800 technical publications and one of the world's most cited physicists. Recipient of numerous awards and distinctions.

Steven Louie
Scientific Advisor
Senior faculty scientist in the Materials Sciences Division at Lawrence Berkeley Laboratory. Professor of Physics at UC Berkeley. Globally recognized expert with h-index over 150.

Laine Conklin
Advisor
President at Conklin Communications. Expert in communications and media coaching for senior executives from startups to Fortune 100.

Marta Bulaich
Advisor
Marketing Executive with over two decades of working in the trenches with venture capital firms (Ridge, Canaan, DFJ) and startups in IT and Healthcare.
Investors
Alchemist Accelerator
Pre-seed
Impulse VC
Pre-seed
Breakout Labs
Pre-seed
Partners

ITOCHU Techno-Solutions Corporation offers a comprehensive service package ranging from platform on-boarding and technical support to helping optimize the product utilization and consulting on specific research topics. More information is here.

Impulse Technology offers our platform to users in the Indian market and can provide technical support and consulting on specific research topics. More information is here.
Contact us
535 Mission street, Suite 2021
San Francisco, CA 94103
+1 (510) 473-7770
info@exabyte.io
Case studies

AI FOR SURFACE CATALYSIS
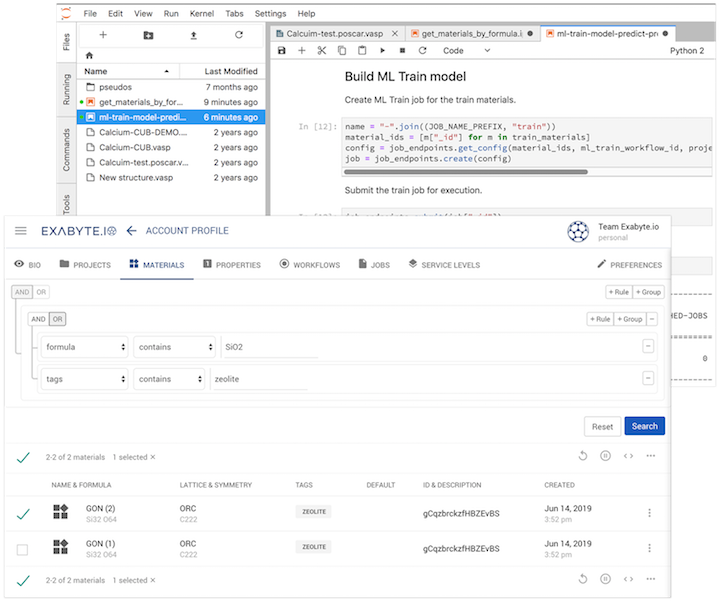
A team of researchers lead by professor Rocca at the Universit'e de Lorraine computed adsorption entalpies in zeolites with high accuracy using the random phase approximation first-principles technique coupled with machine learning.

LIGHT-WEIGHT ALLOYS
A team of researchers from Intermolecular Inc. compressed what would otherwise be 10 years of computing into under 2 days and screened 296 promising structural alloys for potential applications in automotive and aerospace fields.
STRONGER COMPOSITE MATERIALS
Tokyo University of Science team together with ITOCHU Techno-solutions successfully demonstrated a computational evaluation of the mechanical properties of carbon fiber/polymer resin interfaces.

SOLID-STATE BATTERY ELECTROLYTES
A team from San Francisco State University benchmarked Exabyte.io platform against a set of incumbents using atomistic simulations of solid-state lithium electrolytes, and found a 2-3x decrease in timer required to run simulations.
Publications performed on Exabyte.io
Evaluation of interface properties of carbon fiber/resin using the full atomistic model considering the electric charge state
1 Tokyo University of Science, 2 ITOCHU Techno-solutions,
Evaluation of interface strength is important in composite material design such as carbon fiber reinforced plastic. Molecular simulation, which considers aspects such as chemical structure, can be used to evaluate the composite interface strength. Therefore, in this study, the interface energies between graphene, considering the electric charge state, and resin (TriA-X polyimide, DGEBA, triethylenetetramine, vinyl ester, and PA6) were evaluated via molecular dynamics simulation. First, the interface energy and experimentally obtained interface strength were compared. Subsequently, the coefficient of determination R2 was calculated using linear approximations from the interface energy and interface strength. In addition, the correlation coefficient was calculated and showed a high correlation. Based on this data, it was conjectured that a relationship exists between the interface strength and interface energy. Furthermore, the validity of the relationship magnitude between the experimentally obtained interface strength and the interface energy obtained via simulation was evaluated. Moreover, considering graphene oxidation, the interface energies between the resin and three forms of graphene (functionalized with OH, COOH, and O groups) were obtained, and the effect of various oxidation surface treatments of graphene on their corresponding interface strengths was investigated.

Electronic properties of binary compounds with high fidelity and high throughput
1 Exabyte Inc.
We present example applications of an approach to high-throughput first-principles calculations of the electronic properties of materials implemented within the Exabyte.io platform[1, 2]. We deploy computational techniques based on the Density Functional Theory with both Generalized Gradient Approximation (GGA) and Hybrid Screened Exchange (HSE) in order to extract the electronic band gaps and band structures for a set of 775 binary compounds. We find that for HSE, the average relative error fits within 22%, whereas for GGA it is 49%. We find the average calculation time on an up-to-date server centrally available from a public cloud provider to fit within 1.2 and 36 hours for GGA and HSE, respectively. The results and the associated data, including the materials and simulation workflows, are standardized and made available online in an accessible, repeatable and extensible setting.

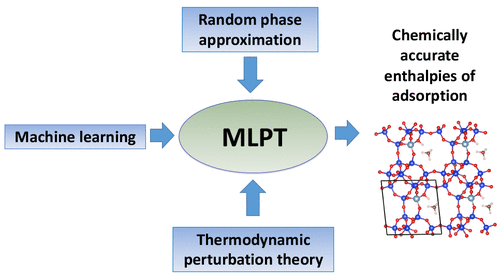
Computing RPA Adsorption Enthalpies by Machine Learning Thermodynamic Perturbation Theory
1 Université de Lorraine, 2 Comenius University in Bratislava, 3 Exabyte Inc.
Correlated quantum-chemical methods for condensed matter systems, such as the random phase approximation (RPA), hold the promise of reaching a level of accuracy much higher than that of conventional density functional theory approaches. However, the high computational cost of such methods hinders their broad applicability, in particular for finite-temperature molecular dynamics simulations. We propose a method that couples machine learning techniques with thermodynamic perturbation theory to estimate finite-temperature properties using correlated approximations. We apply this approach to compute the enthalpies of adsorption in zeolites and show that reliable estimates can be obtained by training a machine learning model with as few as 10 RPA energies. This approach paves the way to the broader use of computationally expensive quantum-chemical methods to predict the finite-temperature properties of condensed matter systems.
Tensile Test Analysis of Carbon Fiber Composite Material by Molecular Dynamics Simulation
1 ITOCHU Techno-solutions, 2 MITSUBISHI GAS CHEMICAL COMPANY, 3 Yokohama National University
In this study, the authors investigate the tensile strength at a carbon fiber/epoxy resin interface using molecular dynamics (MD) simulations. The simulated tension speed and strength were initially estimated, and a realistic tension speed was subsequently selected. The tensile strength calculated using the simulations was in good agreement with the experimental data. Based on the present study and our previous work, the factors contributing to the tensile strength of composite materials were investigated and analyzed. The surface energy between the graphene sheet and resin as well as the molecular structure of the resin in the vicinity of graphene was shown to significantly affect the tensile strength. In addition, the applicability of MD simulation as a useful tool for the prediction and analysis of composite materials was demonstrated. This work used graphene sheets as carbon fiber because it is difficult to express the real carbon fiber for simulation model.
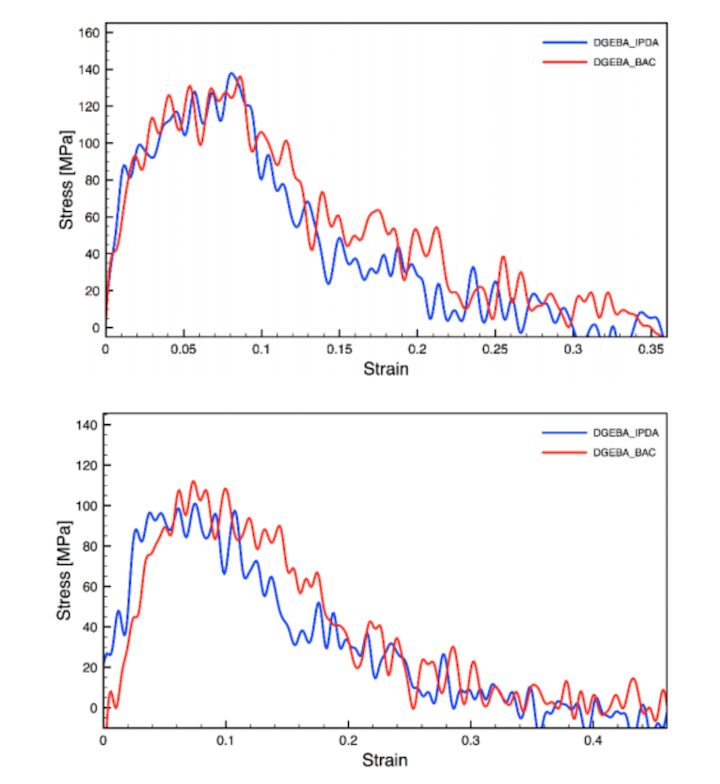
Evaluation of the mechanical properties of carbon fiber/polymer resin interfaces by molecular simulation
1 Tokyo University of Science, 2 ITOCHU Techno-solutions, 3 Japan Aerospace Exploration Agency, 4 Exabyte Inc.,
Herein, we evaluate the mechanical properties of carbon fiber/polymer interfaces using three types of specimens: carbon fiber/vinyl ester resin, carbon fiber/epoxy resin, and carbon fiber/polyimide resin. Microbonding tests were performed and the fiber load at interfacial debonding was obtained. By performing finite element analysis, the true interfacial strengths were determined. The strength values followed the order: polyimide > epoxy > vinyl ester for the specimens tested. Molecular modeling was also performed for these specimens. Three stacked graphene layers were used to represent the carbon fiber surface and the interfacial energy was determined. The interfacial energy of the three systems followed the same order as the strength values. The molecular simulations allowed for a qualitative discussion of the material properties. In addition, an interfacial debonding simulation was per- formed, however the resin part failed instead of the interface, indicating that we evaluated the resin strength but not the interfacial strength. For the quantitative evaluation of interfacial debonding strength, further studies are necessary.

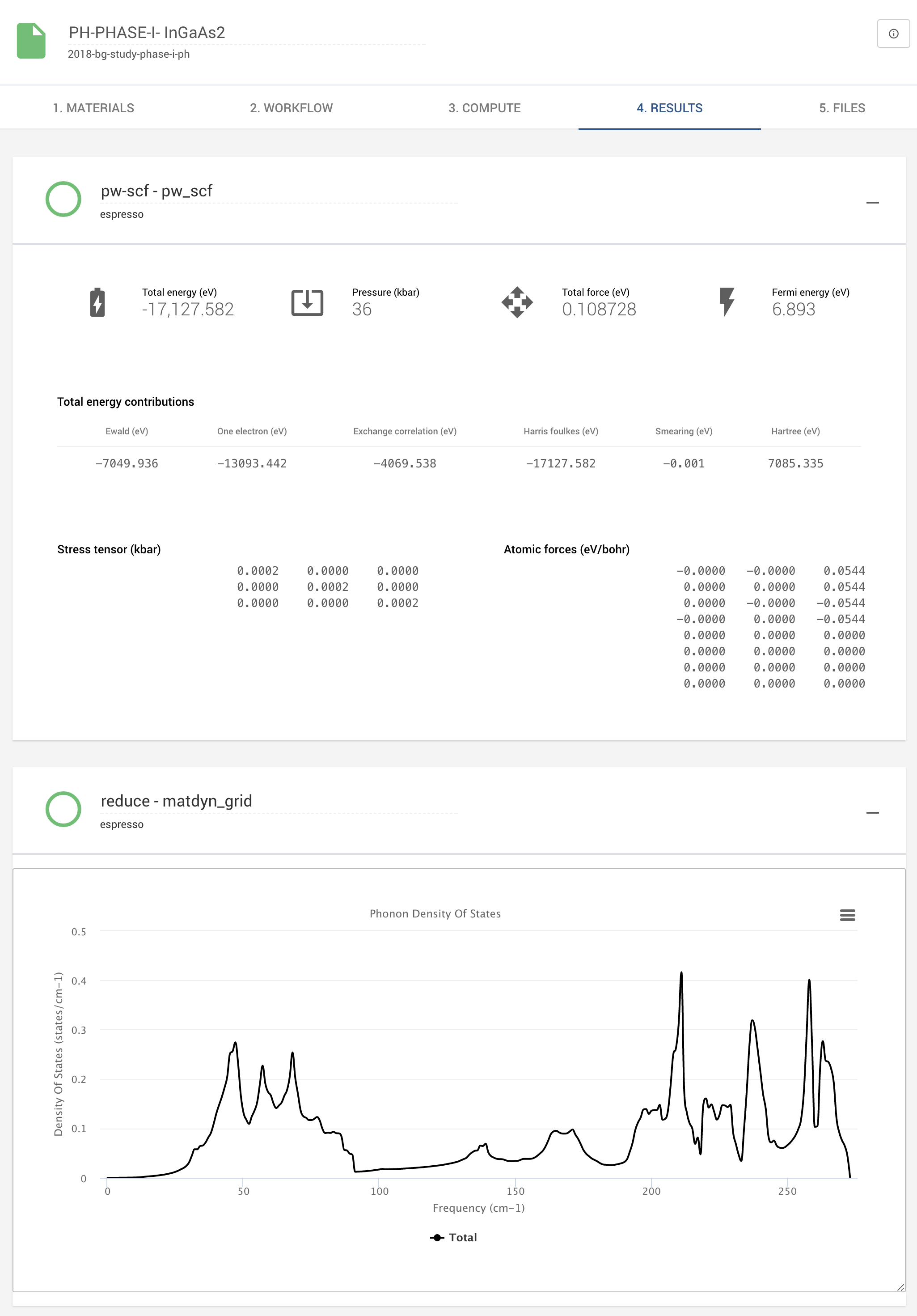
Fast and accessible first-principles calculations of vibrational properties of materials
1 Exabyte Inc.
We present example applications of an approach to first-principles calculations of vibrational properties of materials implemented within the Exabyte.io platform. We deploy models based on the Density Functional Perturbation Theory to extract the phonon dispersion relations and densities of states for an example set of 35 samples and find the results to be in agreement with prior similar calculations. We construct modeling workflows that are both accessible, accurate, and efficient with respect to the human time involved. This is achieved through efficient parallelization of the tasks for the individual vibrational modes. We report achieved speedups in the 10-100 range, approximately, and maximum attainable speedups in the 30-300 range, correspondingly. We analyze the execution times on the current up-to-date computational infrastructure centrally available from a public cloud provider. Results and all associated data, including the materials and simulation workflows, are made available online in an accessible, repeatable and extensible setting.
Accessible computational materials design with high fidelity and high throughput
1 Exabyte Inc.
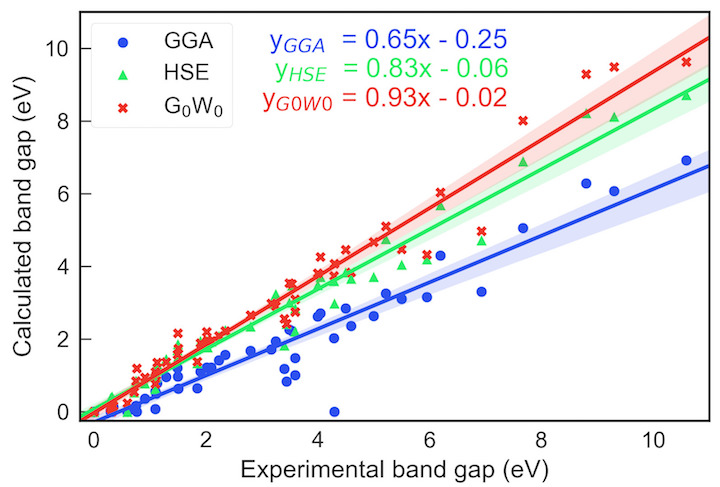
Despite multiple successful applications of high-throughput computational materials design from first principles, there is a number of factors that inhibit its future adoption. Of particular importance are limited ability to provide high fidelity in a reliable manner and limited accessibility to non-expert users. We present example applications of a novel approach, where high-fidelity first-principles simulation techniques, Density Functional Theory with Hybrid Screened Exchange (HSE) and GW approximation, are standardized and made available online in an accessible and repeatable setting. We apply this approach to extract electronic band gaps and band structures for a diverse set of 71 materials ranging from pure elements to III-V and II-VI compounds, ternary oxides and alloys. We find that for HSE and G0W0, the average relative error fits within 20%, whereas for conventional Generalized Gradient Approximation the error is 55%. For HSE we find the average calculation time on an up-to-date server centrally available from a public cloud provider to fit within 48 hours. This work provides a cost-effective, accessible and repeatable practical recipe for performing high-fidelity first-principles calculations of electronic materials in a high-throughput manner.
Prediction of new metastable HfO2 phases: toward understanding ferro- and antiferroelectric films
1 Intermolecular, Inc.
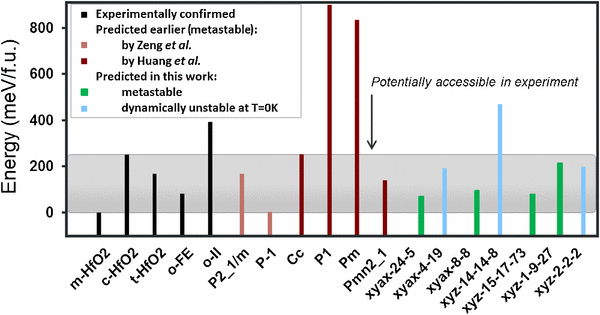
From first principles, we predict several yet-unknown, low-energy, dynamically stable phases of HfO2 . One of the predicted metastable phases has a finite ferroelectric polarization and could be potentially responsible for the ferroelectric and/or antiferroelectric behavior recently reported in thin (Hf,Zr)O2 -based films. Other phases predicted here may potentially form as competing non-ferroelectric phases in thin films, and the possibility of their formation should be taken into account during analysis of experimental thin-film characterization data. These predictions are made possible by an explicit enumeration approach, designed for the case at hand. Our approach outperforms existing theoretical structure prediction methods, including evolutionary algorithms, which have been previously applied to the same problem yet have not identified most of the possible metastable phases found in this study. This suggests that structure enumeration techniques may be indispensable for practical structure prediction problems that seek to identify all low-energy metastable phases rather the single stable (lowest energy) phase.
Publications related to Exabyte.io
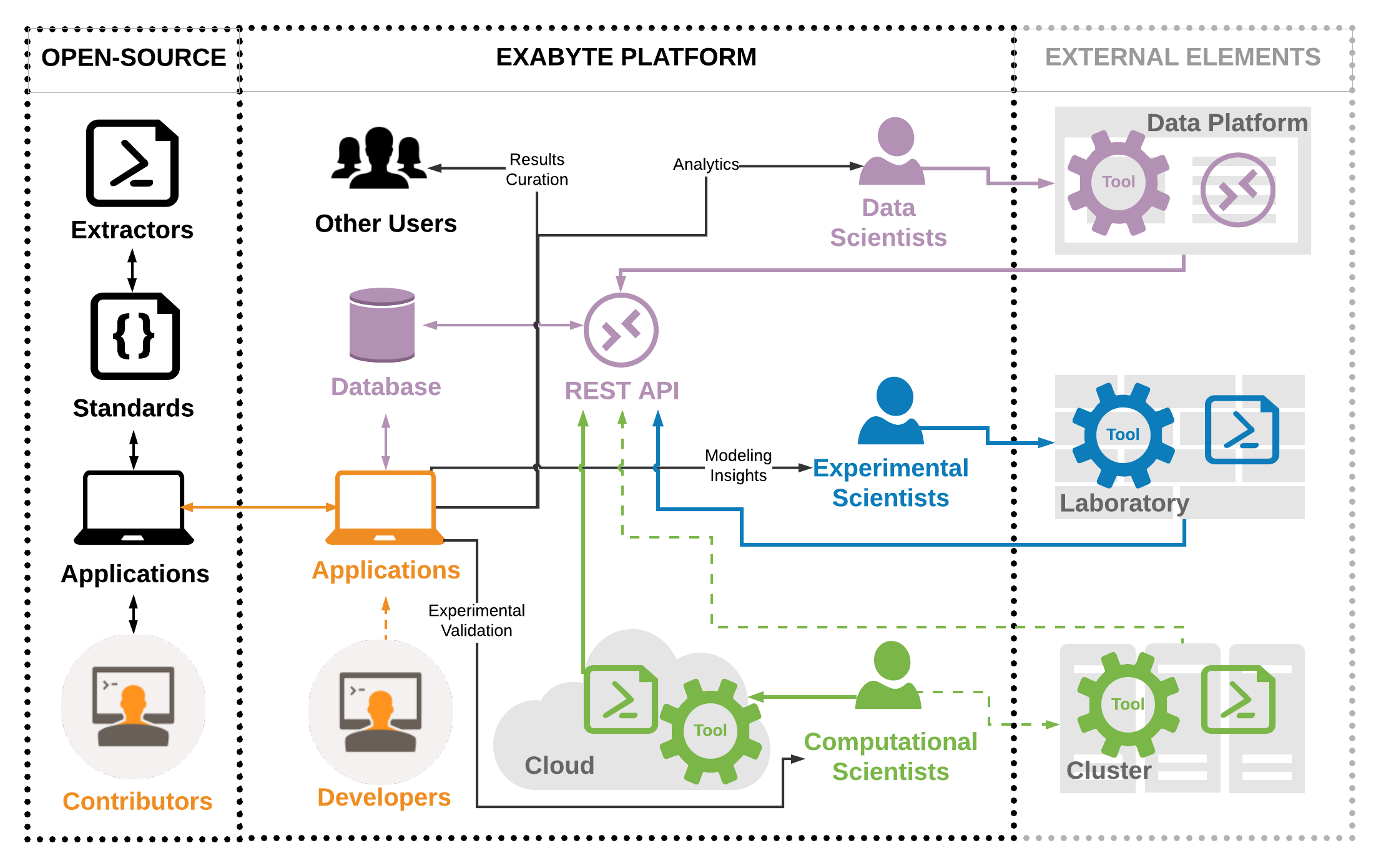
Data-centric online ecosystem for digital materials science
1 Exabyte Inc.
Materials science is becoming increasingly more reliant on digital data to facilitate progress in the field. Due to a large diversity in its scope, breadth, and depth, organizing the data in a standard way to optimize the speed and creative breadth of the resulting research represents a significant challenge. We outline a modular and extensible ecosystem aimed at facilitating research work performed in an accessible, collaborative, and agile manner, without compromising on fidelity, security, and defensibility of the findings. We discuss the critical components of the ecosystem and explain the implementation of data standards and associated tools. We focus initial attention on modeling and simulations from nanoscale and explain how to add support for other domains. Finally, we discuss example applications or the data convention and future outlook.
Continuous evaluation of the performance of cloud infrastructure for scientific applications
1 Exabyte Inc.
Cloud computing recently developed into a viable alternative to on-premises systems for executing high-performance computing (HPC) applications. With the emergence of new vendors and hardware options, there is now a growing need to continuously evaluate the performance of the infrastructure with respect to the most commonly-used simulation workflows. We present an online ecosystem and the corresponding tools aimed at providing a collaborative and repeatable way to assess the performance of the underlying hardware for multiple real-world application-specific benchmark cases. The ecosystem allows for the benchmark results to be stored and shared online in a centrally accessible database in order to facilitate their comparison, traceability, and curation. We include the current up-to-date example results for multiple cloud vendors and explain how to contribute new results and benchmark cases.
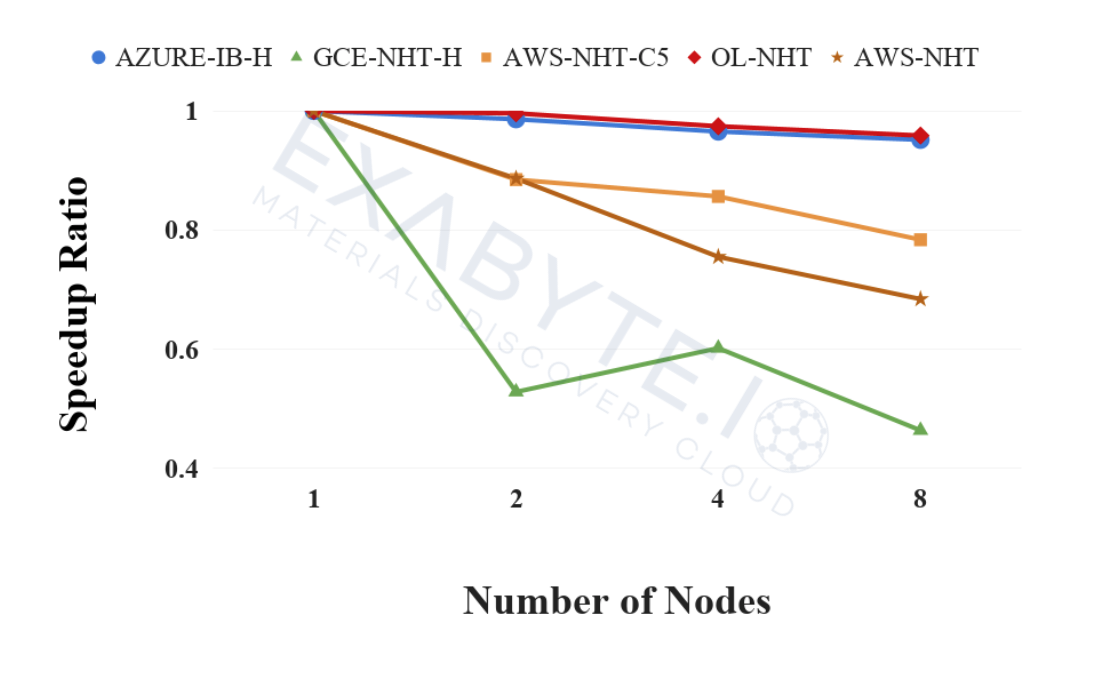
Comparative benchmarking of cloud computing vendors with high performance linpack
1 Exabyte Inc.
We present a comparative analysis of the maximum performance achieved by the Linpack benchmark on compute intensive hardware publicly available from multiple cloud providers. We study both performance within a single compute node, and speedup for distributed memory calculations with up to 32 nodes or at least 512 computing cores. We distinguish between hyper-threaded and non-hyper-threaded scenarios and estimate the performance per single computing core. We also compare results with a traditional supercomputing system for reference. Our findings provide a way to rank the cloud providers and demonstrate the viability of the cloud for high performance computing applications.
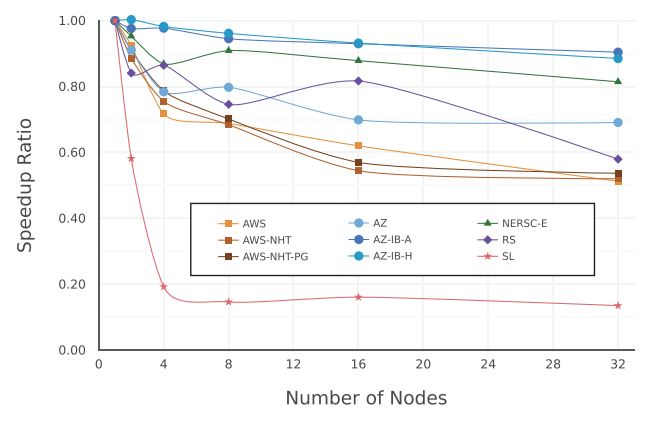
Large-scale high-throughput computer-aided discovery of advanced materials using cloud computing
1 Exabyte Inc. 2 Intermolecular, Inc.
Recent advances in cloud computing made it possible to access large-scale computational resources completely on-demand in a rapid and efficient manner. When combined with high fidelity simulations, they serve as an alternative pathway to enable computational discovery and design of new materials through large-scale high-throughput screening. Here, we present a case study for a cloud platform implemented at Exabyte Inc. We perform calculations to screen lightweight ternary alloys for thermodynamic stability. Due to the lack of experimental data for most such systems, we rely on theoretical approaches based on first-principle pseudopotential density functional theory. We calculate the formation energies for a set of ternary compounds approximated by special quasirandom structures. During an example run we were able to scale to 10,656 CPUs within 7 minutes from the start, and obtain results for 296 compounds within 38 hours. The results indicate that the ultimate formation enthalpy of ternary systems can be negative for some of lightweight alloys, including Li and Mg compounds. We conclude that compared to traditional capital-intensive approach that requires in on-premises hardware resources, cloud computing is agile and cost-effective, yet scalable and delivers similar performance.
Community
Below is a list of institutional affiliations as submitted by users who attempted to register on our platform. This list is included for informational purposes only and has no implication about any potential relationship between our company and the organizations included.
- .
News

Exabyte.io Tapped by USAF to Give Scientists Real-World Version of Tony Stark’s Futuristic Ironman Simulation Software
Exabyte.io, the company unlocking the rapid development of new materials for
Global 2000 companies, announces its selection as a $1M Small Business
Innovation Research (SBIR) Phase II award recipient from the United States Air
Force’s technology incubator, AFWERX. The technology at the center of the award
is Exabyte.io’s Elemental Modeling and Manufacturing Architecture (EMMA), a
real-world version of the futuristic software that Tony Stark used to create new
materials for his “Ironman” suit in the Marvel movie. Exabyte is also actively
hiring to grow its team to support the award and other areas of strong growth.
The AFWERX is a highly competitive program that encourages domestic small
businesses to engage in federal research and research and development with
the potential for commercialization. Exabyte.io received the Phase II award to
conduct research and development and adapt its software to address the application
areas and
materials of interest to the Air Force. The Phase II award was granted after
successfully completing a Phase I evaluation.
“Material requirements for today’s cutting-edge manufacturing are immense
and ever-increasing,” explains Timur Bazhirov, Ph.D., CEO and founder of
Exabyte.io. “This Phase II award underscores the importance of understanding
and exploiting the latest digital R&D methods to keep pace with accelerating
materials and manufacturing lifecycles.”
The Air Force Research Laboratory and AFWERX work together to streamline the
SBIR process and accelerate the development of new technologies, broaden the
pool of potential applicants and decrease bureaucratic overhead. Beginning
in 2018, the Air Force started offering ’Special’ SBIR topics to drive a
broader range of innovations that are faster and leaner.
Released 2020-09-30 via WebWire.
Security and privacy of your data is our first priority. We designed our product to the highest standards of data protection. Below we highlight some of the features that we adopt to keep your data safe.
Confidentiality
Nobody but you can access your data
Encryption in transfer with high-grade SSL/TLS
Encryption at rest with 256-bit AES and kernel-encrypted hard drives
Private simulations on isolated clusters protected by firewall
Secure and comprehensive key management system
Security features inherited from our cloud datacenters
CLOUD PROVIDERS
Availability
Safe backups, another layer of protection
Complete Data backups
performed daily
Redundant highly-available storage system
Distributed high-performant systems architecture
Authentication and Authorization
Secure access and control of user-level permissions
Password complexity
and identity verification
Secure login
Key-based secure
shell authentication
4096-bit encryption
Flexible permission
management scheme
Secure collaboration
Contact us for more information
We support private cloud, on-premises installation, and can
provide details upon request.
The information available here is further explained in our online
documentation available here.